我校动物科学技术学院刘剑锋教授团队在数量遗传学领域全基因组上位效应关联分析方法研究上取得重要进展。他们提出一种快速的全基因组上位效应关联分析方法-REMMA,相对于传统基于单标记混合模型的上位效应关联分析方法,REMMA方法可以更好地控制群体分层和亲缘关系的影响,具有更高的检验效率。相关研究近日在线发表于生物信息学主流期刊《Bioinformatics》上,文章题目为“A Rapid Epistatic Mixed-model Association Analysis by Linear Retransformations of Genomic Estimated Values”,文章链接:https://doi.org/10.1093/bioinformatics/bty017。生物信息学是一门统计学、计算科学与生命科学交叉的前沿学科。在多组学大数据时代,生物信息学理论和计算方法在解析复杂生命过程和现象中发挥关键作用。《Bioinformatics》作为以生物信息学命名的期刊,主要刊登该领域前沿性和方向性研究成果,五年IF 8.044。
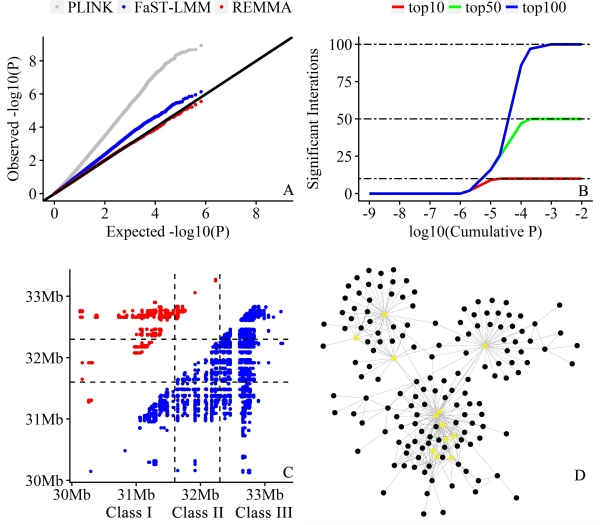
全基因组关联分析是挖掘动植物复杂性状候选基因的重要工具,在这一策略下,大量的功能位点被成功定位,但是这些位点仅能解释很小比例的遗传变异,致使相当比例的遗传力得不到解释,这种现象称为“缺失遗传力”。分析基因间上位效应被认为是进一步挖掘功能突变、解析遗传机制的重要手段。但是计算效率瓶颈限制了全基因组上位效应检验在遗传解析中的实质性应用。传统的基于单标记混合模型检验方法在检验加性效应上有很高的效率,但是应用到上位效应检验上则需要数年计算时间。为了解决上位效应检验效率低的计算难点,刘剑锋教授团队提出了“A Rapid Epistatic Mixed-model Association Analysis”方法,简称REMMA。这种方法同时将加性和上位基因组关系矩阵放入混合模型中,首先估计出个体的上位效应,然后通过线性转换,估计出位点之间的互作效应。相对于传统的单标记混合模型检验方法,REMMA方法具有以下优势:(1)由于同时加入加性和上位基因组关系矩阵,可以更好地控制遗传背景造成的假阳性率;(2)估计位点上位效应的算法复杂度与个体数成线性关系,使全基因组范围内估计位点互作效应成为可能;(3)该方法同样适用于基因组加性和显性效应的估计,在基因组选择遗传评估策略中对亲本后代的表型预测、以及繁殖亲本精准选配具有重要应用价值。 我校刘剑锋教授是论文唯一通讯作者,博士研究生宁超是论文的第一作者,团队成员周磊讲师、加州大学河滨分校的徐士忠教授以及国际家畜研究所的Raphael Mrode教授是本研究的合作作者。
|